Il più grande contributo di un’intelligenza artificiale alla biologia
DeepMind sta pubblicando milioni di previsioni sulla struttura delle proteine: potrebbe avere un impatto enorme nelle scienze della vita
- Condividi
- X
- Regala il Post
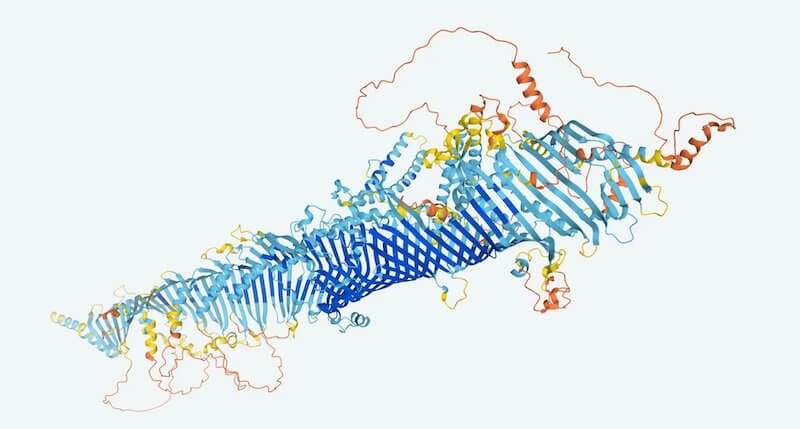
DeepMind, la divisione di Alphabet (Google) che si occupa di intelligenza artificiale (AI), sta realizzando il più grande e completo archivio della struttura delle proteine umane e di alcuni altri esseri viventi, una risorsa che nei prossimi anni potrebbe avere un forte impatto in numerosi ambiti della ricerca, dalle scienze della vita al settore farmaceutico. L’archivio contiene al momento 350mila previsioni sulle caratteristiche e la forma delle proteine, ma l’obiettivo di DeepMind è di arrivare a 100 milioni entro la fine dell’anno.
L’iniziativa è stata resa possibile dai progressi di AlphaFold, un’intelligenza artificiale sviluppata da DeepMind e che già alla fine dello scorso anno aveva dimostrato di essere molto affidabile nel prevedere la forma di una proteina, partendo dai dati sulle catene di amminoacidi che la costituiscono.
Proteine e forma
Per comprendere l’importanza del nuovo archivio occorre fare un rapido ripasso di biologia. Le proteine sono essenziali per la nostra esistenza e per quella degli altri esseri viventi: regolano il metabolismo, la risposta agli stimoli e sono essenziali per il trasporto delle molecole, solo per fare qualche esempio.
Ogni proteina ha proprie caratteristiche determinate dalle catene di amminoacidi che la costituiscono e dalla forma che assume, ripiegandosi su sé stessa. La forma è essenziale nel determinare la funzione, e per questo in biologia molecolare si dice spesso che “la struttura è la funzione” di una proteina.
Buona parte delle proteine ha dimensioni tra 1 e 100 nanometri (un nanometro equivale a un miliardesimo di metro) ed è quindi molto difficile da osservare per studiarne la struttura. Negli ultimi decenni, buona parte degli esperimenti di biologia molecolare ha riguardato proprio le tecniche e i metodi da impiegare per provare a comprendere come specifiche proteine si ripieghino su sé stesse, assolvendo in questo modo a particolari funzioni.
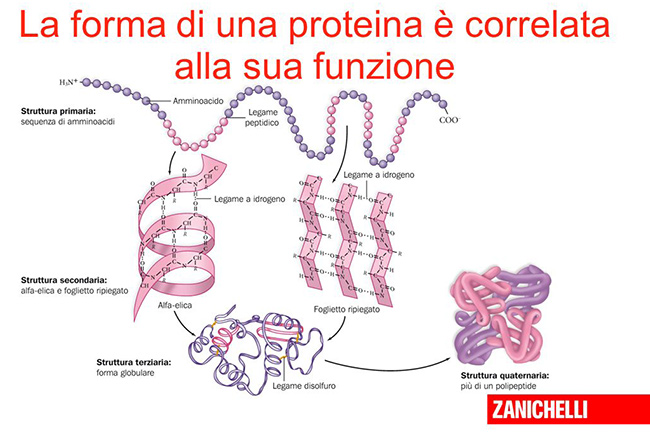
I ricercatori hanno nel tempo sviluppato tecniche che sfruttano i raggi X (cristallografia a raggi X) per osservare la struttura di alcune proteine. Le tecniche si sono poi affinante e nell’ultimo decennio la microscopia elettronica, che prevede l’osservazione dei campioni a temperature estremamente basse, ha offerto migliori risultati e ridotto il rischio di modificare accidentalmente le molecole nella fase di preparazione per essere studiate.
Questo approccio sperimentale richiede comunque molto tempo e macchinari costosi, con lo svantaggio di non portare sempre a risultati soddisfacenti. Nell’infinitamente piccolo, i campioni appaiono come se fossero bidimensionali, con tutte le difficoltà del caso per ricostruire con precisione la struttura tridimensionale della proteina che si sta analizzando.
Previsioni
A partire dagli anni Ottanta, alcuni ricercatori pensarono di risolvere il problema seguendo un approccio diverso: partire dalle catene di amminoacidi e scoprire come queste determinino poi la struttura delle proteine di cui fanno parte (negli amminoacidi non ci sono istruzioni su che forma debbano assumere le proteine: i ripiegamenti sono dovuti alle leggi della fisica). Per farlo fecero ricorso ai computer, che però all’epoca non offrivano grandi capacità di calcolo e la possibilità di avere risultati facilmente riproducibili, uno dei pilastri del metodo scientifico. Accadeva spesso che un modello al computer si rivelasse utile per determinare la struttura di una proteina, ma che fallisse poi miseramente in successivi studi svolti da altri ricercatori.
Le cose sono migliorate negli ultimi 20 anni grazie all’avvento dei sistemi di intelligenza artificiale e al Critical Assessment of Structure Preditction (CASP), una competizione organizzata ogni due anni negli Stati Uniti che premia le migliori soluzioni per prevedere le strutture delle proteine al computer. La sfida consiste nel prevedere una struttura proteica già nota grazie ai classici metodi sperimentali, ma non ancora resa pubblica. Alla fine dello scorso anno, AlphaFold aveva vinto la competizione, producendo una previsione quasi del tutto identica alla struttura ottenuta in precedenza dai ricercatori.
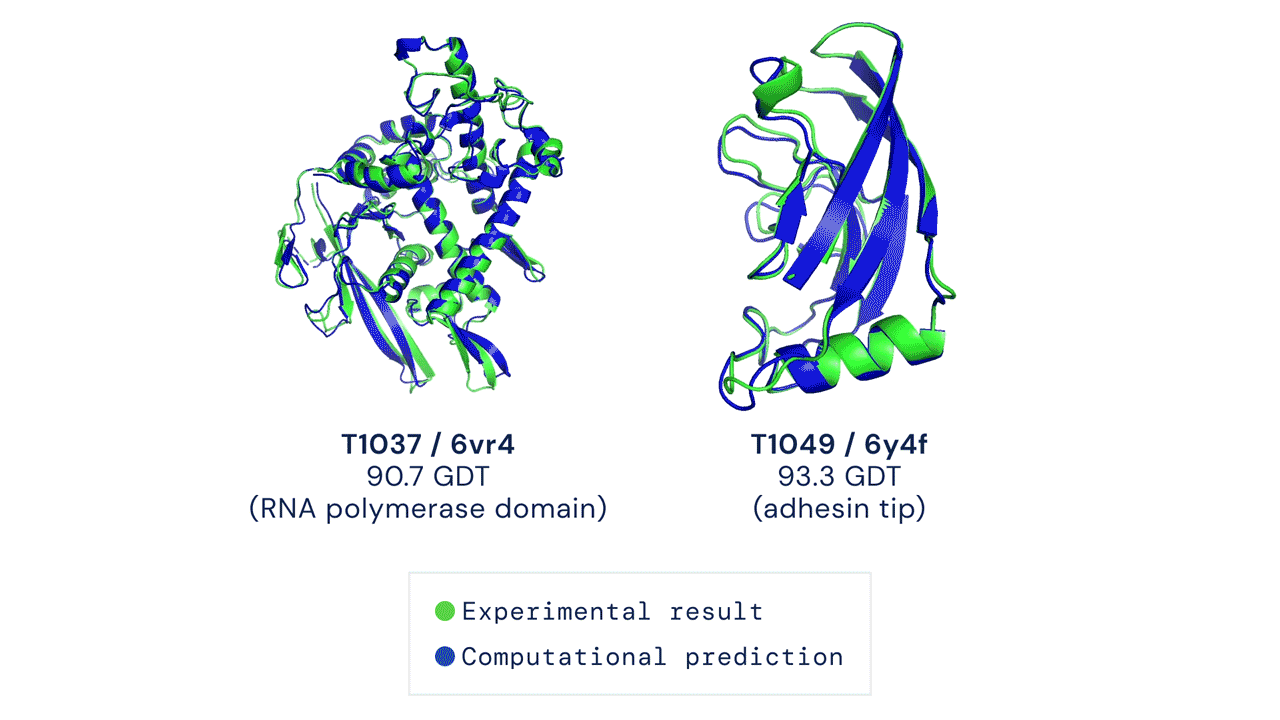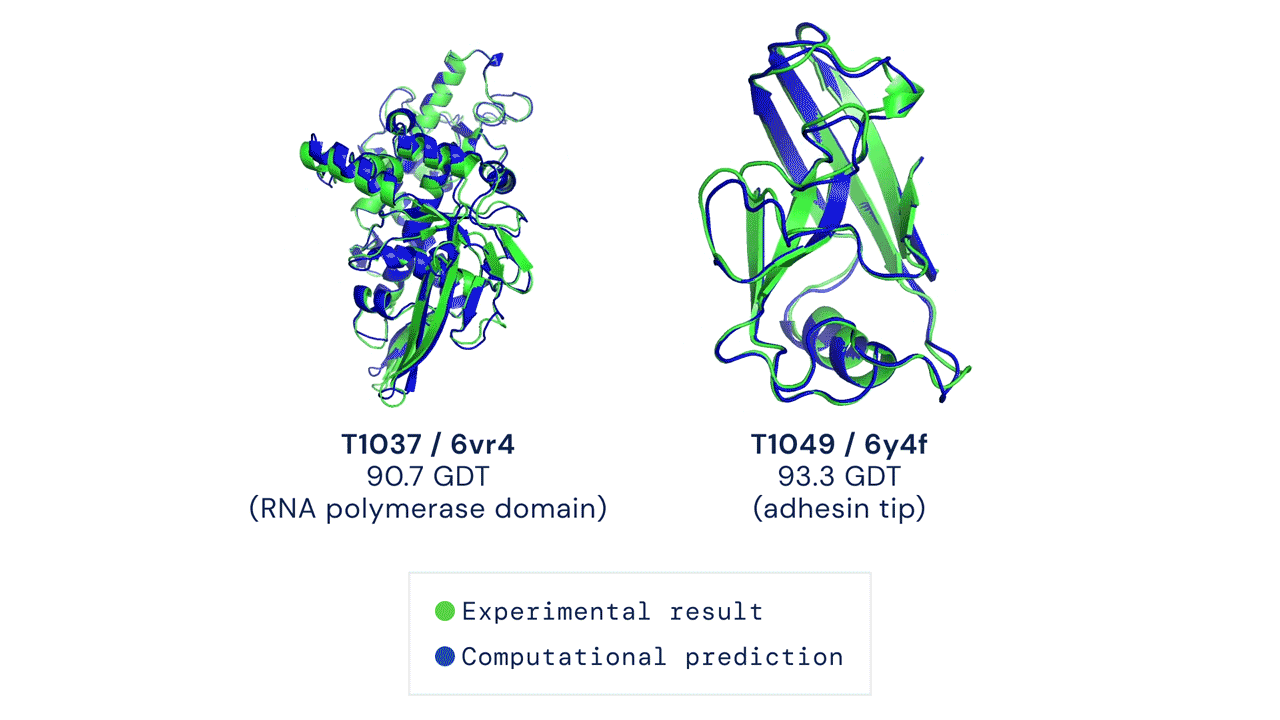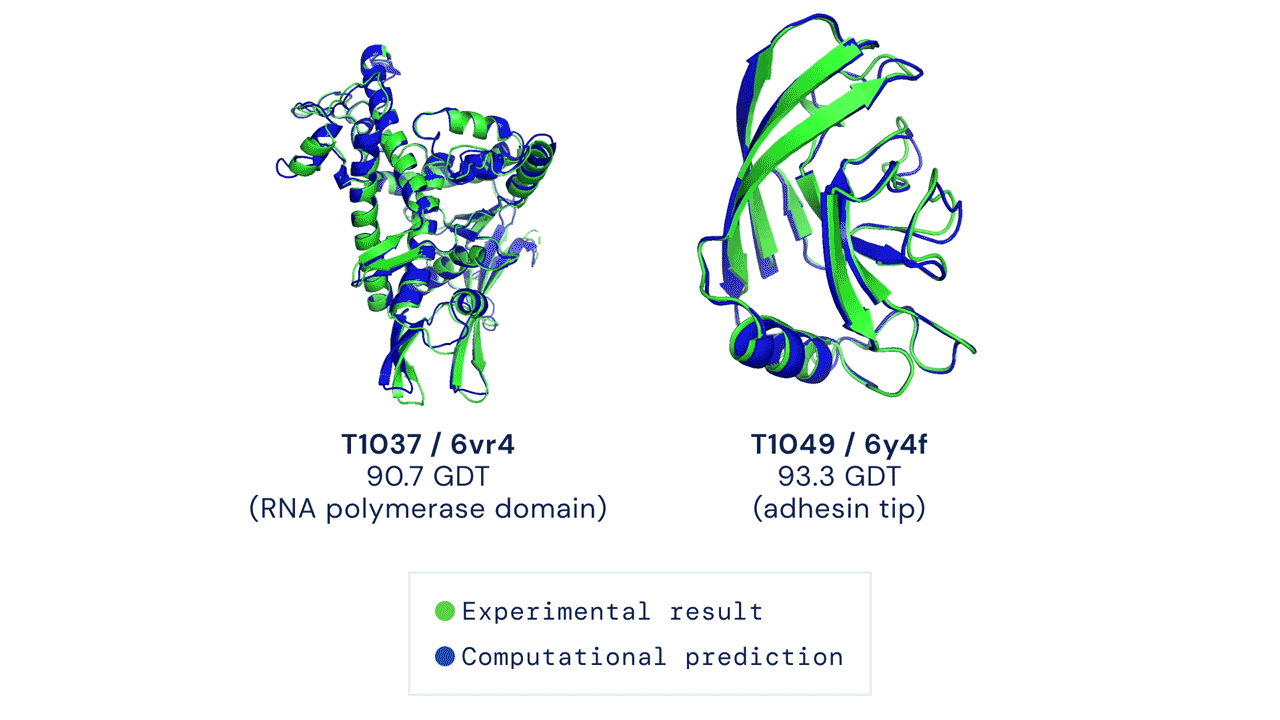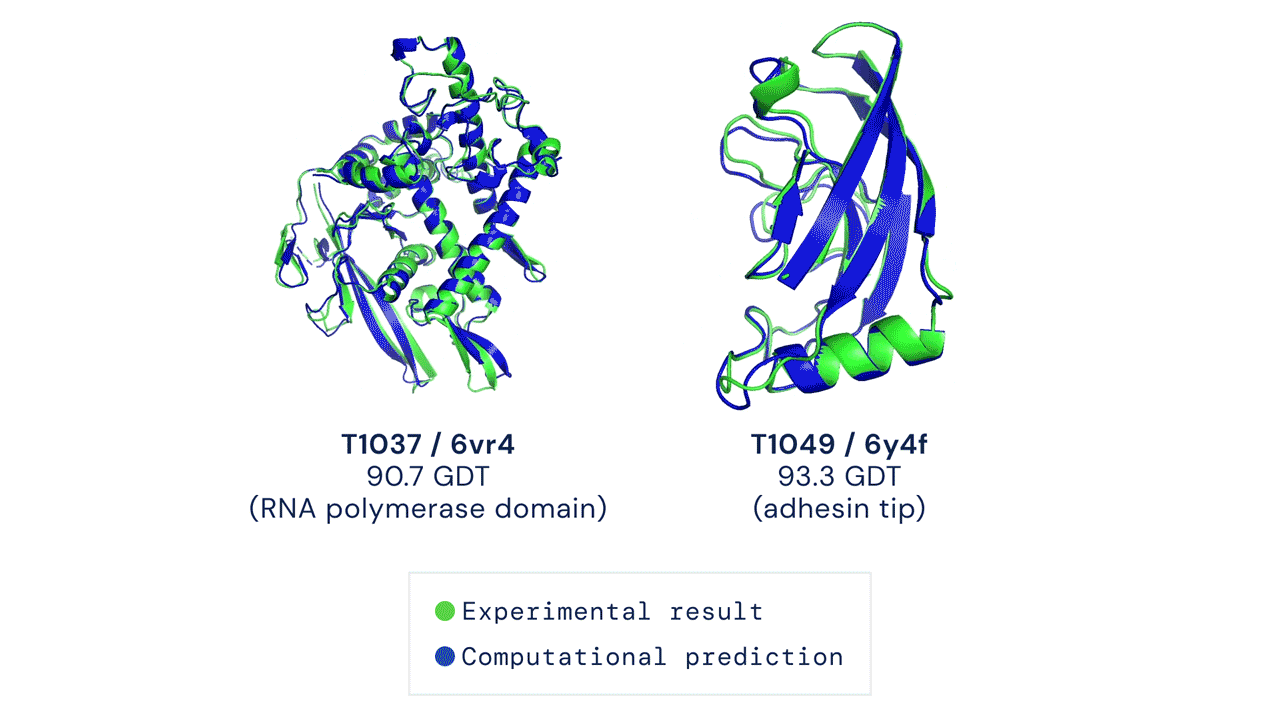
Oltre alla grande accuratezza, AlphaFold aveva dato risultati in pochissimo tempo rispetto ai sistemi concorrenti. Il sistema aveva sorpreso i ricercatori e aveva evidenziato le grandi potenzialità di questa intelligenza artificiale per prevedere con accuratezza la struttura di centinaia di migliaia di proteine, partendo dall’elenco degli amminoacidi che le compongono.
Archivio
Le proteine osservate con metodi sperimentali e finora catalogate sono circa 180mila, disponibili attraverso il Protein Data Bank, uno dei principali archivi per la consultazione delle loro caratteristiche. DeepMind ha messo ora a disposizione 350mila previsioni sulla struttura di altrettante proteine appartenenti a 20 diversi esseri viventi. Per quanto riguarda direttamente la nostra specie, il nuovo archivio comprende la struttura di 20mila proteine, circa il 98 per cento del proteoma umano, il complesso delle proteine che esprime il nostro organismo.
I dati sono liberamente consultabili dalla comunità scientifica tramite un sito dedicato curato dall’European Molecular Biology Laboratory (EMBL), uno dei laboratori più importanti per le scienze della vita. Nei prossimi mesi l’archivio si arricchirà di molte altre previsioni, arrivando a comprendere circa 100 milioni di stime sulla struttura delle proteine. È una mole gigantesca di dati, che secondo gli esperti richiederà anni prima di essere sfruttata completamente dai ricercatori, anche in modi innovativi e difficili da immaginare oggi.
Opportunità
La comprensione della struttura di una proteina è importante per sviluppare nuovi farmaci, progettare enzimi che assolvano a particolari compiti (per esempio per distruggere elementi nocivi per il nostro organismo), o ancora per rendere le piantagioni più resistenti a parassiti e condizioni ambientali estreme, senza dover ricorrere a prodotti chimici. Le informazioni ottenute tramite AlphaFold sono inoltre preziose nella ricerca contro i virus, compreso l’attuale coronavirus.
I ricercatori potranno fare tutte queste cose senza dovere investire tempo e risorse nelle analisi sperimentali per determinare in laboratorio la struttura delle proteine. Potranno lavorare da subito con le strutture previste da AlphaFold, combinandole insieme dove necessario e intervenendo su specifiche caratteristiche. In alcuni casi sarà comunque necessario un successivo lavoro di verifica per assicurarsi che l’AI abbia fatto una previsione affidabile. Questo rischio è comunque mitigato dalla capacità stessa dell’AI di valutare in autonomia la propria previsione, indicando i margini di errore.
La scelta di DeepMind di rendere disponibile per sempre l’intero archivio in forma gratuita è stata accolta molto positivamente dalla comunità scientifica.
Alphabet, la grande holding che controlla Google, spende ogni anno centinaia di milioni di dollari per finanziare le attività di DeepMind per ora in perdita. La società, fondata a Londra nel 2010, fu acquisita da Google nel 2014 e da allora ha sviluppato diversi sistemi di intelligenza artificiale, rivelandosi una delle più promettenti nel settore. Al tempo dell’acquisizione fu concordato che DeepMind potesse proseguire nelle ricerche per favorire nuove importanti scoperte scientifiche, in parallelo con attività più redditizie e che Alphabet potrà sfruttare commercialmente.